Cancer cells are characterized by self-sufficiency in the absence of growth signals, their ability to evade apoptosis, resistance to anti-growth signals, sustained angiogenesis, uncontrolled proliferation, and invasion and metastasis. Alterations in cellular bioenergetics are an emerging hallmark of cancer.
Genome instability, leading to increased mutability, was considered the essential enabling characteristic for manifesting the hallmarks. However, the mutation rate for most genes is low making it unlikely that the numerous pathogenic mutations found in cancer cells would occur sporadically within a normal human lifespan. This then created a paradox. If mutations are such rare events, then how is it possible that cancer cells express so many different types and kinds of mutations? Mutations in each of cancer genes result in dysregulation of metabolic pathways involved in oxygen, iron, energy or nutrient sensing, suggesting that cancer is a disease of cell metabolism.
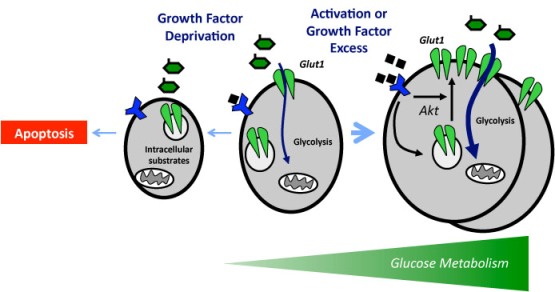
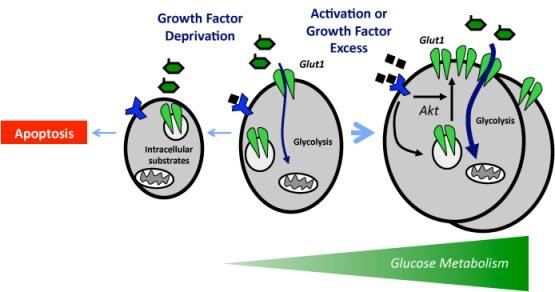
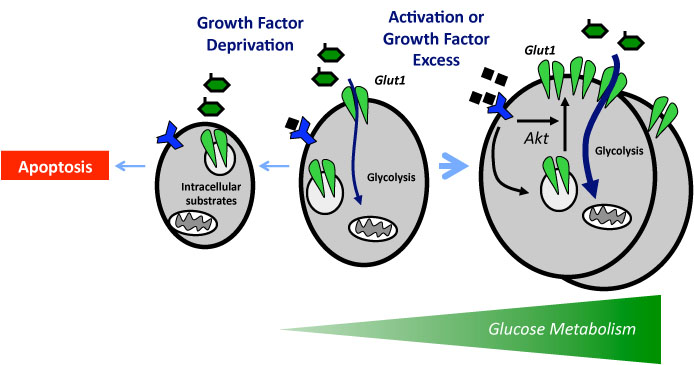
It is critical that cell metabolism be closely linked to cellular energetic and biosynthetic demands. Emerging evidence indicates that impaired cellular energy metabolism is the defining characteristic of nearly all cancers regardless of cellular or tissue origin. In contrast to normal cells, which derive most of their usable energy from oxidative phosphorylation, cancer cells become heavily dependent on substrate level phosphorylation to meet energy demands. As a result of a spectrum of mitochondrial defects, tumor cells often preferentially use glycolysis to generate ATP (adenosine triphosphate), even in the presence of oxygen, a phenomenon known as aerobic glycolysis, or the “Warburg effect.”
The mitochondrion is the major organelle implicated in the cellular bioenergetic and biosynthetic changes accompanying cancer. These bioenergetic modifications contribute to the invasive, metastatic and adaptive properties typical in most tumors. Moreover, mitochondrial DNA mutations complement the bioenergetic changes in cancer. Several cancer management therapies have been proposed that target tumor cell metabolism and mitochondria.
Targeting the fundamental metabolic abnormalities in cancer provides a unique opportunity for the development of more-effective forms of therapy for this disease. Glycolytic inhibitors serve as a classical example of cancer metabolism targeting agents. Several TCA cycle and oxidative phosphorylation inhibitors are being tested for their anticancer potential. Moreover, agents targeting the PDC/PDK (pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase) interaction are being studied for reversal of Warburg effect. Targeting of the apoptotic regulatory machinery of mitochondria is another potential anticancer field in need of exploration. Additionally, oxidative phosphorylation uncouplers, potassium channel modulators, and mitochondrial redox are under investigation for their anticancer potential.
Targeting Glucose Metabolism: An Emerging Concept for Anticancer Therapy.
Manipulation of glycolysis in malignant tumors: fantasy or therapy?
Carbohydrate metabolism via glycolysis and the TCA (tricarboxylic acid) cycle is pivotal for cancer growth, and increased refined carbohydrate consumption adversely affects cancer survival. We know that glucose is the primary substrate for aerobic glycolysis, the primary metabolic pathway in cancer cells. The intake of sugar and sweetened foods was clearly linked to the development of pancreatic and other cancers.
The cells metabolized the sugars differently. While sugar of any kind offered sustenance, fructose played a key role in the proliferation of cancer cells. That means the cancer spread more quickly on a high-fructose diet. If scientists can develop a drug that blocks a cancer cell’s ability to metabolize fructose, they might be able to halt the spread of the disease.
Traditionally, glucose and fructose have been considered as interchangeable monosaccharide substrates that are similarly metabolized, and little attention has been given to sugars other than glucose. However, fructose intake has increased dramatically in recent decades and cellular uptake of glucose and fructose uses distinct transporters. All sugars are not created equal.
Fructose induces transketolase flux to promote pancreatic cancer growth.
Sugar, in the form of glucose, activates the IKK beta enzyme and promotes the development of diabetes via an enhanced inflammatory response. IKK beta is required for activation of a protein called nuclear factor kappa B (NF-kB), that acts as a master switch to turn on inflammation in response to bacterial or viral infections. In epithelial cells, NF-kB promotes the development of cancer not through inflammation, but through inhibition of a cell-killing process called apoptosis. In myeloid cells, NF-kB causes the expression of pro-inflammatory molecules that stimulate the division of genetically altered epithelial cells and thereby increase tumor size.
The IkappaB kinase – a bridge between inflammation and cancer.
NF-kappaB and cancer: how intimate is this relationship.
Glucose activates NF-kB signaling which promotes inflammation. We know that most cancers originate from sites of chronic inflammation. The followings are excellent studies which explain, in detail, how glucose increases NF-kB activity. The study shows how a mixed meal stimulates ROS (reactive oxygen species) generation and increases NF-kB activity.
The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages.
The activation of NF-kB is known to promote resistance to both chemo drugs and radiation. When a person undergoes chemo or radiation and consumes a carbohydrate rich diet, there is a strong probability that this diet will contribute to the failure of their treatment protocol. Fortunately, salicylates and sulfasalazine bind directly to the IKK beta enzyme thereby inhibiting its ability to activate NF-kB signaling. The following study provides information on oral dosages of aspirin (sodium salicylate). The subjects were treated with 7 grams of aspirin daily for two weeks. Their glucose disposal rate and insulin resistance improved remarkably. This was attributed to the inhibition of IKKbeta (NF-kB) enzyme activity by salicylates. In addition to improving glucose metabolism, salicylates also prevent fat induced insulin resistance.
Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes.
The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta.
Prevention of fat-induced insulin resistance by salicylate.
Sulfasalazine is the only drug that was developed specifically for arthritis and combines a sulfa drug with acetylsalicylic acid. Sulfasalazine is used primarily as an anti-inflammatory agent in the treatment of inflammatory bowel disease (IBD) as well as for rheumatoid arthritis. The following studies identify sulfasalazine as a direct inhibitor of IKK-alpha and -beta by antagonizing adenosine triphosphate binding. The suppression of NF-kB activation by inhibition of the IKKs contributes to the well-known anti-inflammatory and immunosuppressive effects of sulfasalazine.
Sulfasalazine inhibits activation of nuclear factor-kappaB in patients with ulcerative colitis.
The IkappaB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus.
Certain cancers depend for growth on uptake of cystine/cysteine from their environment. Sulfasalazine is a potent inhibitor of the x(c) (-) cystine transporter. Sulfasalazine-induced cystine/cysteine starvation leading to glutathione depletion may be useful for therapy of certain cancers dependent on extracellular cystine. These effects were shown to result primarily from inhibition of cystine uptake mediated by the x(c) (-) cystine transporter and not from inhibition of NF-kB (nuclear factor kappaB) activation, another anticancer property of sulfasalazine.
Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-kappaB.
Sulfasalazine-induced cystine starvation: potential use for prostate cancer therapy.



{kind=link}
{kind=link}
{kind=link}